
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| 26 | 27 | 28 | 29 | 30 |
- quantum chemistry
- Gaussian
- 양자역학
- 화학
- Chemistry
- 물리화학
- Aromaticity
- 여행
- 타이베이
- 유기화학
- travel
- 양자화학
- Computational Chemistry
- Physical Chemistry
- Organic Chemistry
- 대만
- Today
- Total
Daily Project
[Gaussian] Single Point Energy Calculation #3 본문
[Gaussian] Single Point Energy Calculation #3
Jun_Hyeong 2020. 12. 15. 20:13
#Gaussian Output File
이전 포스팅1에서 Job script와 output 파일에서 정보를 얻어오는 방법에 대해서 알아보았습니다. 이를 이용해서 Formaldehyde의 Single point Energy Calculation을 진행하고 output 파일에서 얻을 수 있는 정보에 대해서 알아보겠습니다.
- Ground State Energy
먼저, Formaldehyde의 바닥 상태 에너지를 알아봅시다. log 파일에서 SCF Done 키워드를 이용해서 특정 분자의 바닥상태 에너지를 얻을 수 있습니다.
|
SCF Done: E(RB3LYP) = -114.495058176 A.U. after 11 cycles
NFock= 11 Conv=0.66D-08 -V/T= 2.0094
|
cs |
이때, 에너지의 단위가 A.U. 임을 주의해야 합니다. A.U. 은 atomic unit이라는 뜻으로 hartree를 의미합니다. 1 hartree ≈ 2625.5 kJ/mol ≈627.51kcal/mol ≈27.21eV가 성립합니다. 예를 들어, 특정 분자의 흡수 파장(absorption wavelength)을 계산할 때, 바닥 상태 에너지와 들뜬 상태 에너지의 차이를 구하고 hartree를 적절한 단위(eV)로 변환시킨 후 파장(nm)으로 변환시켜줄 수 있습니다.
Guassian 계산을 통해 에너지를 비교할 때 가장 중요한 것은 분자를 구성하는 원자의 종류와 수가 모두 동일 하면서 동일한 화학 모델(Model Chemistry)을 사용했을 때 비교 가능하다는 것입니다. 만약, Formaldehyde와 Acetone에 대해서 Single point Energy Calculation를 진행해서 에너지를 얻었을 때, 이를 비교하는 것은 의미가 없습니다.
- Dipole Moment and Higher Multipole Moment
두 번째로, Dipole moment와 Multipole moment에 대한 정보를 알아봅시다. 먼저, Dipole moment는 적용된 전기장에 대한 에너지의 1차 도함수로 분자 전하 분포의 비대칭 성을 알려줍니다. 이는 log 파일에서 Dipole moment 키워드를 이용해서 정보를 얻을 수 있습니다.
|
Dipole moment (field-independent basis, Debye):
X= 0.0000 Y= 0.0000 Z= -2.4254 Tot= 2.4254
Quadrupole moment (field-independent basis, Debye-Ang):
XX= -11.4472 YY= -11.2990 ZZ= -11.7954
XY= 0.0000 XZ= -0.0000 YZ= -0.0000
Traceless Quadrupole moment (field-independent basis, Debye-Ang):
XX= 0.0667 YY= 0.2149 ZZ= -0.2816
XY= 0.0000 XZ= -0.0000 YZ= -0.0000
Octapole moment (field-independent basis, Debye-Ang**2):
XXX= -0.0000 YYY= -0.0000 ZZZ= 0.8089 XYY= -0.0000
XXY= -0.0000 XXZ= 0.8049 XZZ= -0.0000 YZZ= -0.0000
YYZ= -0.3128 XYZ= -0.0000
Hexadecapole moment (field-independent basis, Debye-Ang**3):
XXXX= -9.3016 YYYY= -16.8327 ZZZZ= -45.0193 XXXY= -0.0000
XXXZ= 0.0000 YYYX= -0.0000 YYYZ= -0.0000 ZZZX= 0.0000
ZZZY= 0.0000 XXYY= -4.5880 XXZZ= -9.0018 YYZZ= -9.9694
XXYZ= 0.0000 YYXZ= 0.0000 ZZXY= -0.0000
|
cs |
Dipole moment 아래에 있는 X와 Y, Z는 Dipole moment의 벡터 성분을 나타내며, Tot는 Dipole moment의 크기를 의미합니다. 즉, Formaldehyde의 경우 C=O를 중심으로 대칭이기 때문에 Dipole moment가 Z축 성분만이 존재하는 것입니다.
Quadrupole은 적용된 전기장에 대한 에너지의 2차 도함수로 분자 모양에 대한 간단한 정보를 제공합니다. 예를 들어, 특정 분자가 동일한 XX와 YY, ZZ의 값을 가진다면 분자의 전하 분포는 구형을 가진다는 것을 의미합니다. Formaldehyde의 경우 XX와 YY, ZZ가 서로 대략적으로 비슷한 값을 가지고 있습니다. 그러나, ZZ가 XX와 YY에 비해서 조금 더 큰 값을 가지고 있는 사실을 통해 전하 분포가 전체적으로 구형을 가지면서 Z 축 방향으로 elongation이 있을 것을 예측할 수 있습니다.

- Molecular Orbital
마지막으로 Formaldehyde의 분자 오비탈을 얻어보겠습니다. 생성된 chk 파일을 formchk 명령어를 이용해서 fchk 파일로 변환하고 GaussView로 파일을 불러옵니다.

fchk 파일이 열리면 Result 탭의 Surfaces/Contours를 클릭하면 아래와 같은 새로운 창이 뜹니다.
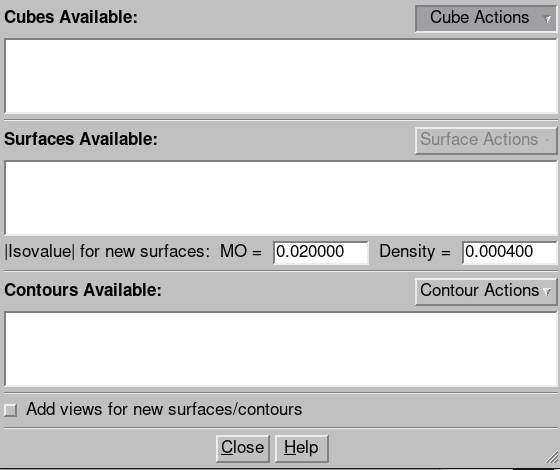
Cubes Available 칸의 Cube Actions을 클릭하고 New Cube를 선택하면 여러가지 Cube를 생성할 수 있는데, Type을 Molecular Orbital로 지정하고 Orbitals를 HOMO, LUMO로 설정하면 HOMO와 LUMO에 해당하는 Cube가 생성됩니다.

그 후, 원하는 Cube를 선택 후 Surfaces Available 칸의 Surface Action을 클릭하고 New Surface를 선택하면 원하는 분자 오비탈에 해당하는 Surface가 생성됩니다. Surface Action에서 표시하거나 숨길 수 있으며, Figure 5는 이 방법을 통해 얻은 Formaldehyde의 HOMO와 LUMO입니다. Type을 변경하면 Figure 1과 같은 전하 분포를 얻을 수도 있습니다.


또는, GaussView의 MO를 누르고 Visualize를 클릭한 후, Add Type을 HOMO, LUMO로 지정한 후 Update 버튼을 눌러주는 방법으로 분자 오비탈을 생성할 수 있습니다.

이 포스팅은 Exploring Chemistry with Electronic Structure Methods, Third Edition를 기반으로 합니다.
'Chemistry Project > Gaussian' 카테고리의 다른 글
| [Gaussian] Single Point Energy Calculation #2 (0) | 2020.12.04 |
|---|---|
| [Gaussian] Single Point Energy Calculation #1 (6) | 2020.11.23 |
| [Gaussian] Introduction to Gaussian #2 (0) | 2020.11.22 |
| [Gaussian] Introduction to Gaussian #1 (0) | 2020.10.21 |



